
Eine große Zahl von Einzelnukleotid-Varianten (Single Nucleotide Variants, SNVs) wurde in den letzten Jahren unter Nutzung von sogenannten DNA-Arrays bei nahezu sämtlichen Probanden (insgesamt ca. 8000) der populationsbasierten Gesundheitsstudie Vorpommern (Study of Health in Pomerania, SHIP; Völzke et al., 2011;PubmedArtikel) typisiert. Gegenwärtig ist es unter Nutzung der so generierten SNV-Typisierungsdaten möglich, für jedes Probanden-Genom den allelischen Zustand bzw. Genotyp von ca. 17 Millionen SNVs zu bestimmen.
In genetischen Assoziationsstudien werden diese SNV-Daten mit in SHIP erhobenen Phänotyp-Variablen in Beziehung gesetzt. Dies ermöglicht die Identifizierung von SNV-Allelen, die statistisch signifikant mit definierten Variablen assoziiert sind, und damit von genetischen Loci, deren kodierte Information die jeweiligen Phänotypen beeinflusst. Meist sind die primär detektierten SNV-Allele nicht diejenigen, die den spezifischen Phänotypen kausativ zu Grunde liegen, und eine nachfolgende Feinkartierung muss jene erst identifizieren. Dennoch liefern genetische Assoziationsstudien wertvolle Hinweise auf Varianten im Kontext von Kandidaten-Genen, die Einfluss auf die Ausprägung von Phänotypen haben.
Verantwortliche Mitarbeiter
Wissenschaftliche Mitarbeiter
Dr. Stefan Weiß
Spezifische Forschungsprojekte
Der Schwellenwert für die sogenannte Genom-weite Signifikanz liegt per definitionem bei p=5x10-8, das heißt, dieser p-Wert muss für eine Assoziation zwischen SNV-Allel und Phänotyp erreicht oder unterschritten werden, damit diese als Genom-weit signifikant kategorisiert wird. Der Wert von 5x10-8 definiert sich historisch durch die Annahme von 1x106 unabhängigen genetischen Loci im humanen Genom (Pe'er et al., 2008; pmid: 18348202; doi: 10.1002/gepi.20303). Der nominelle Signifikanz-Schwellenwert von 0,05 muss daher entsprechend für multiples Testen nach Bonferroni korrigiert werden (0,05/106=(5×10−8).
Die mittels moderner Typisierungs-Arrays erzeugten SNV-Datensätze erfassen für sogenannte häufige SNVs (Common Variants) die Variation im gesamten humanen Genom, wobei Common Variants durch eine Häufigkeit des selteneren der beiden Allele eines SNVs (die Minor Allele Frequency, MAF) von mehr als 5 % in der entsprechenden Referenz-Populations-Stichprobe definiert sind. Die aktuellste Referenz-Datenbank für MAFs ist dabei jene, die auf dem 1000 Genomes Project basiert, in dem die Genome von 2.500 Individuen unterschiedlicher ethnischer Herkunft sequenziert wurden. Genetische Assoziationsstudien, die auf solchen Typisierungs-Datensätzen beruhen, werden folglich als Genom-weite Assoziationsstudien bezeichnet (Genome-wide Association Studies, GWAS). Unvollständiger wird die Variation im Genom für intermediär häufige Varianten (Low-Frequency Variants, 5 % ≥ MAF≥ 0,5 %) und insbesondere für seltene Varianten (Rare Variants, MAF< 0,5 %) mittels der Array-basierten Typisierung erfasst. Daraus folgt, dass in Array-basierten GWASs primär solche Genotyp-Phänotyp-Assoziationen detektierbar sind, die auf Common Variants beruhen.
Seit dem Jahr 2009 ist die Abteilung für Funktionelle Genomforschung an GWAS beteiligt, wobei eine Fülle unterschiedlicher Phänotypen adressiert wurde. Die Anzahl der daraus resultierenden, mehrheitlich hochrangigen Publikationen liegt mittlerweile bei weit über 100. Um eine maximale statistische Power zu erreichen, werden GWAS heute mehrheitlich in großen internationalen Konsortien durchgeführt, was es ermöglicht, sehr große Zahlen typisierter Individuen in die Studien einzuschließen, häufig im Bereich einiger 100.000. Dies erlaubt es, auch schwächere Assoziationen Genom-weit signifikant zu detektieren. Zwei Beispiele für derartige internationale Konsortien, an denen auch die Abteilung für Funktionelle Genomforschung mitwirkt, sind CHARGE (Cohorts for Heart and Aging Research in Genomic Epidemiology; www.chargeconsortium.com) mit thematischen Schwerpunkten im Bereich der Herz-Kreislauf- sowie der Alternsforschung sowie SpiroMeta mit einem Fokus auf Analysen zur Lungenfunktion. Innerhalb der Konsortien werden die Ergebnisse der einzelnen Studien-Kohorten –wie SHIP – in sogenannten Meta-Analysen gemeinsam ausgewertet.
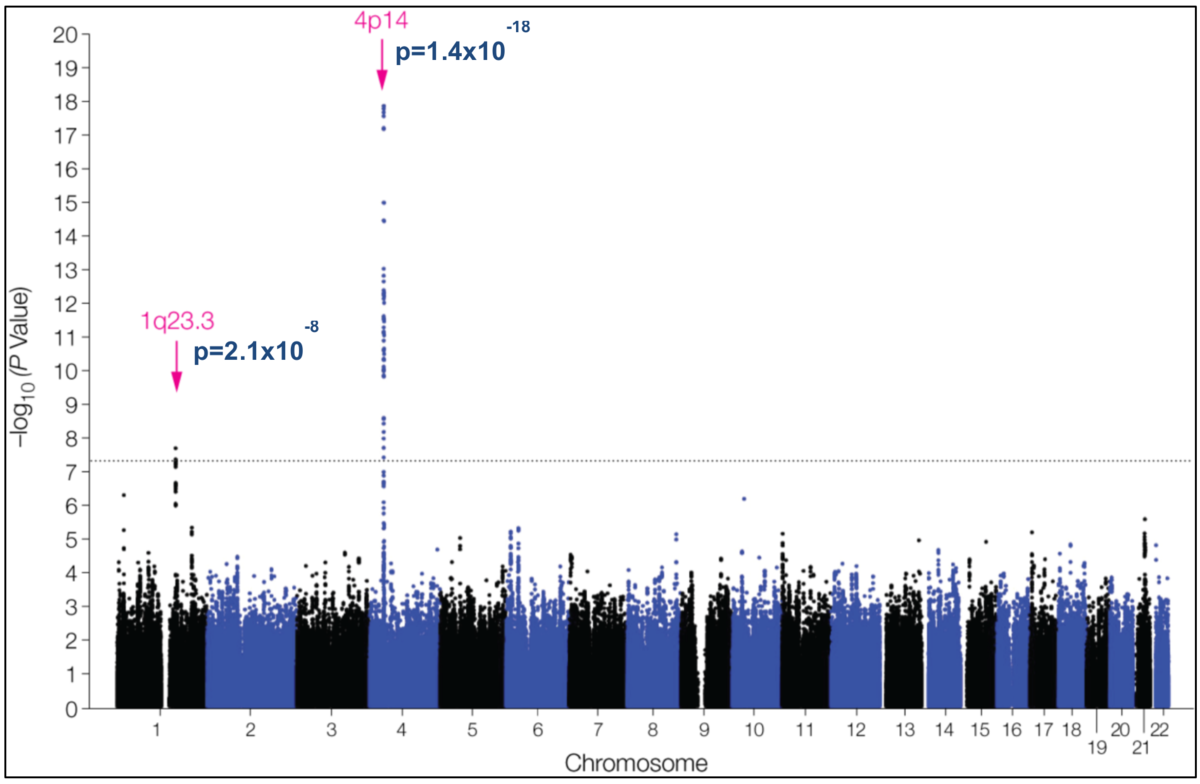
Abbildung 1: Die Ergebnisse von GWAS werden typischerweise in sogenannten Manhattan-Plots dargestellt. Das hier gezeigte Beispiel illustriert das Ergebnis einer GWAS-Meta-Analyse zum Phänotyp „Anti-Helicobacter pylori-IgG-Titer“. Dabei wurden die Ergebnisse entsprechender Serum-Antikörper-Bestimmungen von ca. 4000 SHIP-Probanden sowie ca. 7000 Probanden der ähnlich gestalteten niederländischen Rotterdam Study kombiniert und mit Array-basierten SNV-Typisierungsdaten in Beziehung gesetzt. Auf der Abszisse sind die Chromosomen (mit Ausnahme der Heterosomen) angeordnet, auf der Ordinate der negative dekadische Logarithmus der Wahrscheinlichkeitswerte (P-Werte), mit denen ein SNV-Allel mit dem Phänotyp assoziiert ist. Jeder Punkt des Plots repräsentiert eine Variante. Je höher ein Punkt folglich liegt, desto signifikanter ist die Assoziation zwischen der entsprechenden SNV und dem Anti-Helicobacter pylori-IgG-Titer. Der Phänotyp wurde als sogenannte dichotome (zweiteilige) Variable definiert, wobei jene 25 % der Probanden mit den höchsten Antikörper-Titern mit den restlichen 75 % verglichen wurden. Analysiert wurden nur SNVs mit einer MAF> 1 % in der Gesamt-Stichprobe. Die GWAS-Meta-Analyse ergibt, dass SNVs im Bereich zweier genetischer Loci auf dem langen Arm von Chromosom 1 und dem kurzen Arm von Chromosom 4 Genom-weit signifikant mit dem Anti-Helicobacter pylori-IgG-Titer assoziiert sind (aus Mayerle et al., 2013; doi: 10.1001/jama.2013.4350; pmid: 23652523).
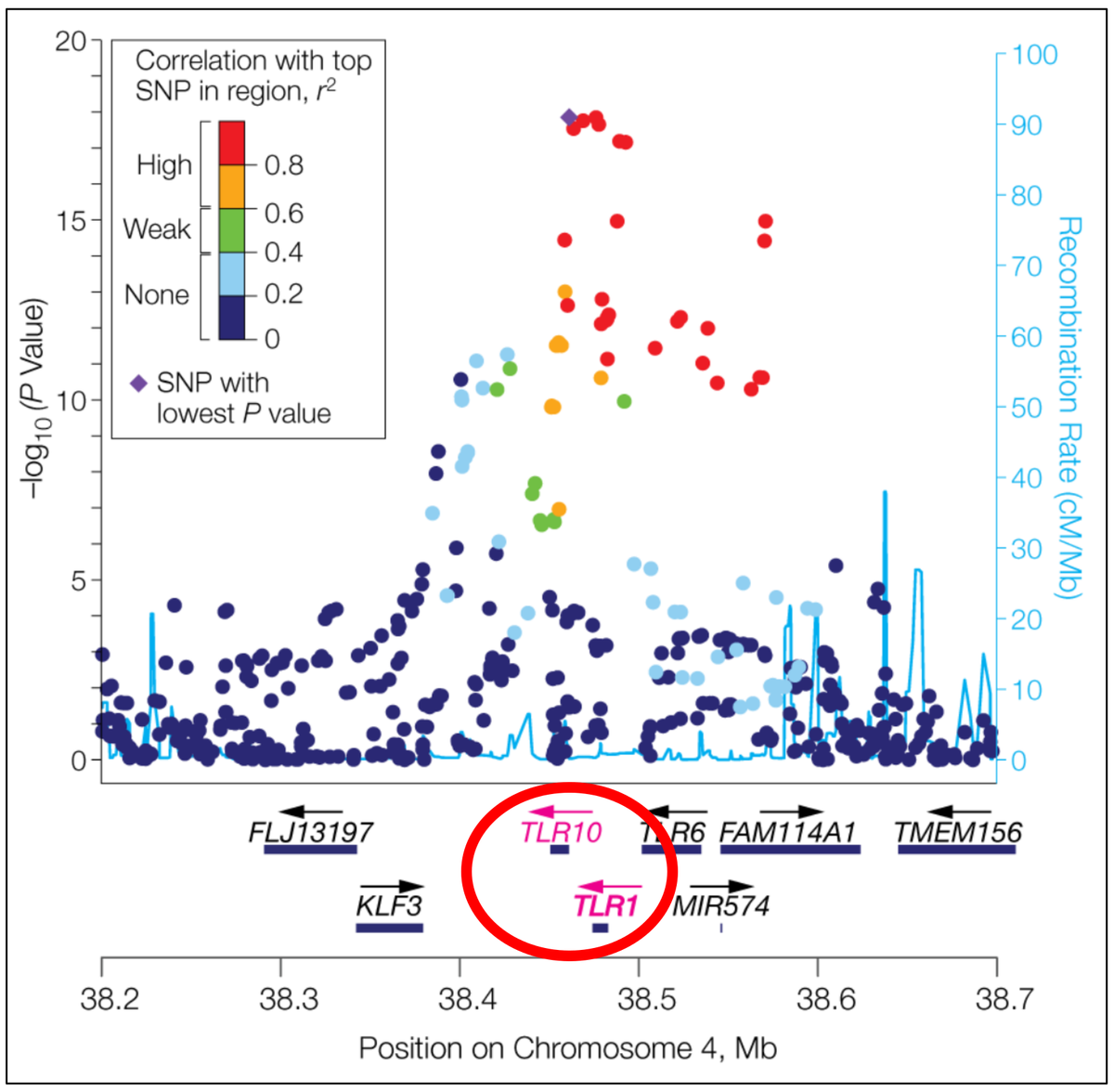
Abbildung 2: Eine weitere typische Form der Darstellung von GWAS-Ergebnissen ist der sogenannte Regional Association Plot. Dabei wird quasi in einen Manhattan-Plot „hineingezoomt“. Im hier dargestellten Beispiel wurde der chromosomale Lokus 4p14 aus dem Manhattan-Plot in Abbildung 1, in dessen Bereich Genom-weit signifikant mit dem Phänotyp „Anti-Helicobacter pylori-IgG-Titer“ assoziierte SNVs detektiert wurden, höher aufgelöst. Auf der Abszisse ist nun der Chromosomenbereich mit den zugehörigen Koordinaten dargestellt, außerdem die dort lokalisierten Gene, deren Transkriptionsrichtung durch Pfeile veranschaulicht wird. Auf der linken Ordinate findet sich wiederum der negative dekadische Logarithmus der Wahrscheinlichkeitswerte (P-Werte), mit denen ein SNV-Allel mit dem Phänotyp assoziiert ist, auf der rechten Ordinate die ermittelte lokale meiotische Rekombinationsrate, angegeben in Centimorgan pro Megabase DNA, die im Plot als hellblaue Linie visualisiert ist. Die am stärksten mit dem Phänotyp assoziierte SNV (oder SNP, Single Nucleotide Polymorphism) ist als lila Rhombus hervorgehoben, die anderen Varianten sind entsprechend ihrer Korrelation mit derselben entsprechend der Legende oben links farbkodiert. Es zeigt sich, dass die stärkste SNV-Phänotyp-Assoziation im Bereich der beiden Gene TLR10 und TLR1 auszumachen ist, die deshalb ebenfalls farblich herausgehoben sind. Beide Gene kodieren Rezeptoren der angeborenen Immunabwehr, sogenannte Toll-like-Rezeptoren, die in die Erkennung bakterieller Antigene involviert sind (aus Mayerle et al., 2013; doi: 10.1001/jama.2013.4350; pmid: 23652523).
Die in den großen GWAS der letzten Jahre detektierten Assoziationen erklären letztlich nur einen geringen Teil der Erblichkeit der analysierten Phänotypen, der sich typischerweise im niedrigen einstelligen Prozentbereich der aus Zwillings- und anderen Verwandtschaftsstudien abgeleiteten Gesamt-Erblichkeit bewegt. In diesem Zusammenhang wurde der Begriff der Missing Heritability, der fehlenden Erblichkeit, geprägt. Es stellt sich die Frage, wie diese zu erklären ist. Ein Ansatz hierzu beruht auf der Tatsache, dass die bislang durchgeführten GWAS weit überwiegend auf Common Variants basierten, die mittels der verwendeten Arrays typisierbar waren, während seltenere Varianten damit nicht erfasst wurden. Häufige Varianten zeichnen sich jedoch durch nur geringe Effektstärken aus. Andererseits geht aus verschiedenen populationsgenetischen Modellen hervor, dass der humane Genpool namentlich im letzten Jahrtausend aufgrund der dramatischen Zunahme der Weltbevölkerung mit seltenen Varianten angereichert sein sollte, die große Effektstärken besitzen (Keinan and Clark, 2012; doi: 10.1126/science.1217283; pmid: 22582263). Diese Varianten, für welche die bislang durchgeführten „klassischen“ GWAS blind waren, könnten demnach die Missing Heritability zumindest teilweise erklären und einen besonders hohen Stellenwert im Kontext der Identifizierung krankheitsrelevanter genetischer Determinanten einnehmen.
Solche Überlegungen trugen wesentlich zum Design einer neuen Klasse von Typisierungs-Arrays bei, den sogenannten Exom-Arrays, womit wiederum nahezu sämtliche 8000 Probanden der SHIP-Kohorte typisiert wurden. Der für SHIP verwendete Exom-Array ermöglicht zwar keine Abdeckung der Genom-weiten Variabilität für Common Variants, jedoch gehören 85 % der ca. 250 000 mit ihm typisierbaren SNVs mit einer MAF< 1 % zur Kategorie der selteneren Varianten, zudem spezifizieren ca. 91 % der SNVs veränderte Proteinsequenzen (daher der Name Exom-Array) und mehrere 1000 weitere Varianten bekannte funktionelle Modifikationen regulatorischer Elemente wie Promotoren oder mRNA-Spleiß-stellen. Ein großer Teil der mit dem Exom-Array typisierbaren Varianten wurde im 1000 Genomes Project identifiziert. Es besteht die Hoffnung, in Assoziationsstudien unter Nutzung der mit dem Exom-Array generierten Typisierungsdaten solche Varianten zu identifizieren, die sich insbesondere durch große Effektstärken bei geringer Häufigkeit in der Population auszeichnen, wie von den erwähnten populationsgenetischen Modellen vorhergesagt. Für Exom-Array-basierte Assoziationsstudien ist die Zusammenarbeit in großen Konsortien in noch höherem Maße essentiell, da nur dadurch die erforderliche Stichproben-Größe für die seltenen Varianten erreicht werden kann, die – auch im Falle größerer Effektstärken – für statistische Analysen erforderlich ist. Zu bedenken bleibt dabei jedoch, dass mittels des auch für SHIP verwendeten Exom-Arrays lediglich 250 000 der 76 Millionen Low-Frequency- und Rare Variants typisiert werden können, die im 1000 Genomes Project identifiziert wurden.
Seit dem Jahr 2014 ist die Abteilung für Funktionelle Genomforschung in mehreren Konsortien auch an auf Exom-Array-Typisierungsdaten basierenden genetischen Assoziationsstudien zu einer Vielzahl verschiedener Phänotypen beteiligt, die bislang in mehreren hochrangigen Publikationen ihren Niederschlag gefunden haben.
Für insgesamt ca. 1000 SHIP-Probanden besteht weiterhin die Möglichkeit der Durchführung Transkriptom-weiter Assoziationsstudien (TWAS). Hierzu wurde aus Vollblut-Proben der Probanden Total-RNA präpariert, die anschließend für Array-basierte Transkriptom-Analysen eingesetzt wurde. Damit konnten die relativen Häufigkeiten sämtlicher in der jeweiligen Probe enthaltenen Transkripte bestimmt werden, die mittels der Arrays detektierbar sind. Diese lassen sich – analog zu genetischen Assoziationsstudien - mit Phänotypen in Beziehung setzen und auf statistische Signifikanz prüfen. Signifikante Assoziationen weisen auf entsprechende physiologische Zusammenhänge hin, wobei stets bedacht werden muss, in wie weit derartige Vollblut-Transkript-Profile auf andere Gewebe/ Organe übertragbar sind, da sie ja ausschließlich auf den Genexpressionsprofilen von Hämozyten basieren. Entsprechende Analysen werden durch die Abteilung für Funktionelle Genomforschung seit dem Jahr 2012 durchgeführt, auch hier vielfach im Rahmen größerer Konsortien.
Erwähnt werden muss schließlich die Mitwirkung der Abteilung für Funktionelle Genomforschung bei der Durchführung von genetischen Assoziationsstudien und TWAS, die nicht auf SHIP-Daten basieren, sondern auf der Patienten-basierten GANI_MED-Kohorte (GANI_MED: Greifswald Approach to Individualized Medicine). Im GANI_MED-Projekt wurden und werden mehrere Patientenkohorten rekrutiert: eine kardiale Kohorte (Herzinsuffizienz), eine zerebrovaskuläre Kohorte (Apoplexie) und eine nephrologische Kohorte (Niereninsuffizienz) sowie eine Komorbiditätskohorte (Metabolisches Syndrom), außerdem eine Kohorte an einem noch nicht vollständig etablierten Krankheitsbild (Fettleber) (Grabe et al., 2014; pmid: 24886498; doi: 10.1186/1479-5876-12-144). Insgesamt ca. 4.000 GANI_MED-Patienten wurden bereits Array-basiert SNV-typisiert, und für 1.200 zusätzlich Vollblut-Transkriptom-Daten erhoben.
Im Rahmen sämtlicher dargestellter Assoziationsstudien war bzw. ist die Abteilung für Funktionelle Genomforschung maßgeblich an der Präparation und Qualitätskontrolle der analysierten Bioproben (DNA, RNA) aus Probanden- bzw. Patientenmaterial, an der Qualitätskontrolle und Prozessierung der generierten Array-Primärdaten sowie an den eigentlichen statistischen Analysen beteiligt, sowie in einer Reihe von Fällen an der physiologischen Interpretation der Assoziationsbefunde.